
S4 (Andarine): The Complete Guide
*DISCLAIMER: Selective Androgen Receptor Modulators (SARMs) are not approved by the FDA for any currently acceptable medicinal/therapeutic purpose. Therefore, this article does not serve as medical advice and is not to be intended to be used to diagnose, treat, cure or prevent any disease. SARMS are to be used for research purposes only and not intended for human consumption.*
What is S4?
S4, also known as Andarine, is considered to be one of the first members of the class of drugs known as selective androgen receptor modulators (SARMs). S4 was originally developed for the treatment of muscle-wasting conditions (cachexia) and benign prostatic hyperplasia (BPH); the latter of which is the noncancerous growth of the prostate.
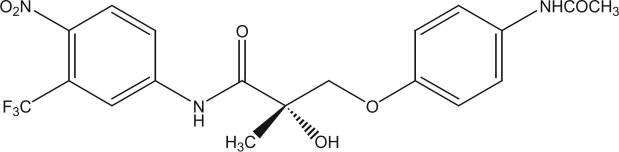
S4 is developed by GTX, Inc.; the same company that created the SARM known as Ostarine, or Enobosarm. In fact, S4 is technically Ostarine’s predecessor. Despite what appeared to be promising preclinical data for S4, research on it was abandoned in favor of its successor Ostarine. While the rationale behind this was never made explicit, one can postulate this was due to Ostarine’s ability to produce more efficacious results in a safer manner compared to S4.
It is nonsteroidal, orally bioavailable, and is selective for anabolic actions in skeletal muscle and bone tissue. The latter prevents androgenic side effects that are common with androgen-based therapies, such as male-pattern baldness (androgenic alopecia) and BPH.
Mechanism of Action
S4 has been proven to be tissue-selective in preclinical research, stimulating anabolic organs more than androgenic organs [1]. Simply put, “anabolic” refers to muscle-building, whereas “androgenic” refers to the development of male sex characteristics [2].
Compared to testosterone therapy, S4 is not as potent in terms of its androgenic activity. But its anabolic activity is similar and sometimes even greater than testosterone [1]. S4 also did not cause any significant amounts of follicle-stimulating hormone (FSH) or luteinizing hormone (LH) suppression, unlike testosterone. Suppression of these hormones most often indicates inhibition of endogenous testosterone production, as these hormones are heavily involved in testosterone synthesis.
S4 has been shown to have a remarkably high bioavailability, which is the amount of the drug that is actually absorbed by the body versus the amount that is excreted either through urine, feces, or some other medium. It has a 95% rate of bioavailability and is rapidly absorbed into the bloodstream only 48-84 minutes after oral administration [3].
However, its notably high rate of bioavailability appears to come with a drawback. S4 tends to clear the body at a very rapid rate, giving the drug a half-life of only 4 hours [3]. However, due to the fact that there is no clinical data on humans to support this, it can’t be said that a half-life this short in duration would occur in humans. Anecdotal evidence suggests that this is unlikely the case, especially as recreational doses are exponentially higher than those used during preclinical trials.
Preclinical Trials
S4 vs. Testosterone
The levator ani muscle, a group of 3 muscles located within the pelvis, is often used as a measure of anabolic activity in preclinical research involving anabolic and/or androgenic compounds. Below is an illustration of its anatomy in humans:
![]()
At a dose of 0.3mg/day in castrated rats, the muscle weight of the levator ani muscle group was fully restored to that of the weight of rats in the non-castrated control group [1]. At the same time, prostate weight was only restored to 33.8% compared to the control, whereas testosterone treatment increased prostate weight to 121% to that of the control group. This illustrates S4’s ability to be selective towards the target organ of interest without promoting unwanted androgenic effects that traditional androgen therapy (in this case testosterone therapy) is notorious for causing.
![]()
This was representative of some of the most convincing SARMs data during the time in which this study was published (2003).
S4 vs. Dihydrotestosterone (DHT)
Another study had compared S4 to dihydrotestosterone (DHT), the highly androgenic and potent derivative of testosterone.
The conversion of testosterone to DHT occurs via the 5α-reductase enzyme. DHT binds to the androgen receptor (AR) at ~2-3x greater affinity than testosterone [4]. In other words, it binds to the AR with double to triple the strength that testosterone normally would. As a result, DHT has the propensity to be 10x more potent as a result.
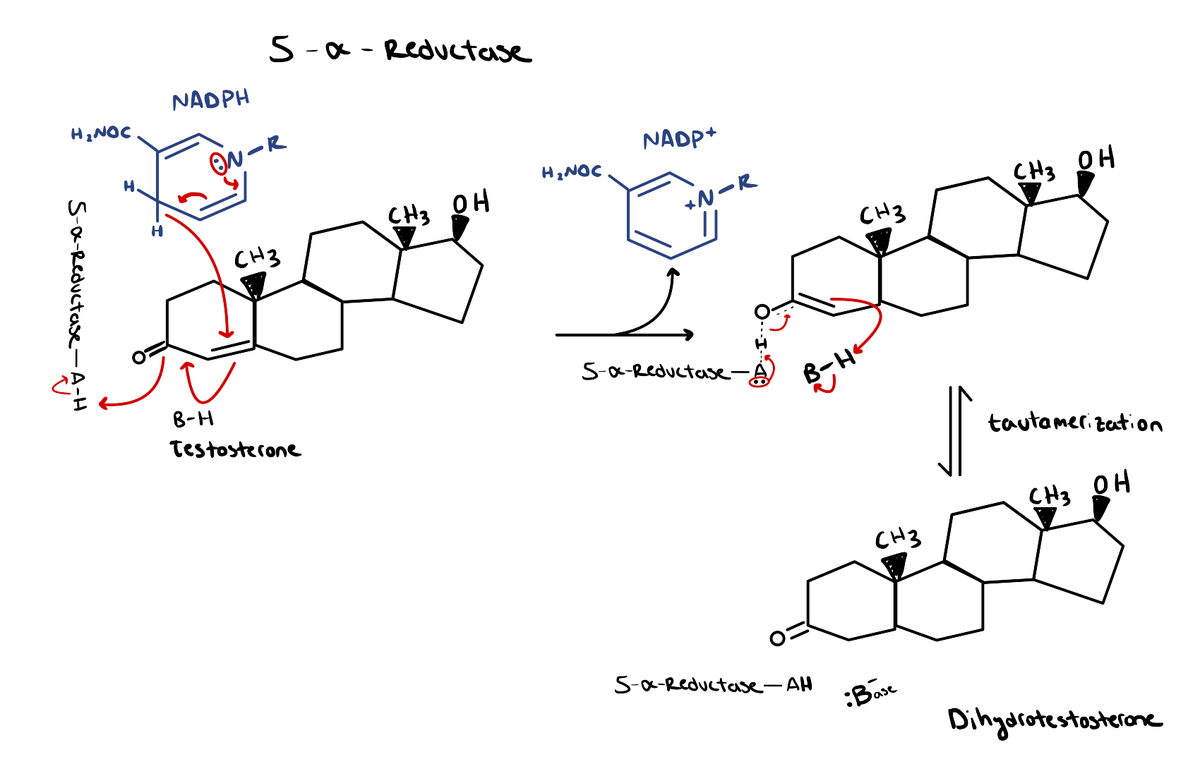
DHT is instrumental in the development of male sexual characteristics during childhood and adolescence, such as facial/body hair, penis/testicle maturation, and prostate growth. During adulthood, though, it plays more of a pathological role.
DHT-related conditions can be related to both abnormally high or low levels of DHT in the body. Conditions that commonly result from excessively high DHT levels are male-pattern baldness (androgenic alopecia) and prostate enlargement, the latter of which can cause BPH and prostate cancer.
Low levels of DHT would cause ultimately cause a state of androgen deficiency, resulting in the onset of various undesirable symptoms, such as:
- Loss of libido
- Infertility
- Decreased muscle mass
- Increased fat mass
- Fatigue
- Anxiety/Depression
In the study that compared S4 to DHT, castrated rats were treated with either 3mg/kg or 10mg/kg of S4 or DHT for 12 weeks. At the end of the 12 weeks, the S4 treated rats in both dose groups were able to restore the overall muscle mass of the levator ani (like with the previously discussed S4 study) in addition to restoration of the soleus muscle (the inner calf muscle).
While the DHT-treated rats also experienced the same results, the weights of their prostate and seminal vesicles (which contribute to semen secretion) increased by more than 2x that of the control group. In contrast, the S4-treated rats only had their prostate and seminal vesicle weights increase by 16% and 17%, respectively.
![]()
![]()
Benign Prostatic Hyperplasia (BPH)
Benign Prostatic Hyperplasia (BPH), known more simply as prostate enlargement, is the noncancerous growth of the prostate. It is often associated with undesirable symptoms and complications such as trouble urinating, bladder stones, urinary tract infections, and kidney issues [5].
The most common methods for treatment of BPH involve two classes of drugs; either anti-androgens or 5α-reductase enzyme inhibitors. Anti-androgen drugs (e.g. hydroxyflutamide) directly block any activity at the AR whereas 5α-reductase enzyme inhibitors (e.g. finasteride) inhibit the conversion of testosterone to DHT.
There was a study conducted that compared two SARMs; S1 and S4, to testosterone treatment. S4 again demonstrated its previously established ability to restore levator ani weight while also illustrating reductions in prostate and seminal vesicle weight [6].
![]()
However, S1 proved to be superior in the latter, causing greater reductions in prostate and seminal vesicle weight compared to S4. Because of this, only S1 succeeded in advancing to a direct comparison to the common BPH treatments of hydroxyflutamide and finasteride. S1 was equally effective at lowering prostate weight while possessing a greater ability to maintain muscle mass.
How Does S4 Compare to Other SARMS?
Ostarine (Enobosarm)
Both S4 and Ostarine were developed by the same company GTX Inc. However, research on S4 was abandoned without explicit explanation in favor of Ostarine. Not only that, but there is far more data on Ostarine (especially in human trials) compared to S4. Therefore, it’s not possible to make direct comparisons between the 2 drugs regarding their effects on humans.
As previously mentioned, it can be speculated that Ostrarine demonstrated overall efficacy and safety compared to S4. Although there are no official clinical trials available on S4, there is preclinical data that was conducted on mice that compared S4 to Ostarine.
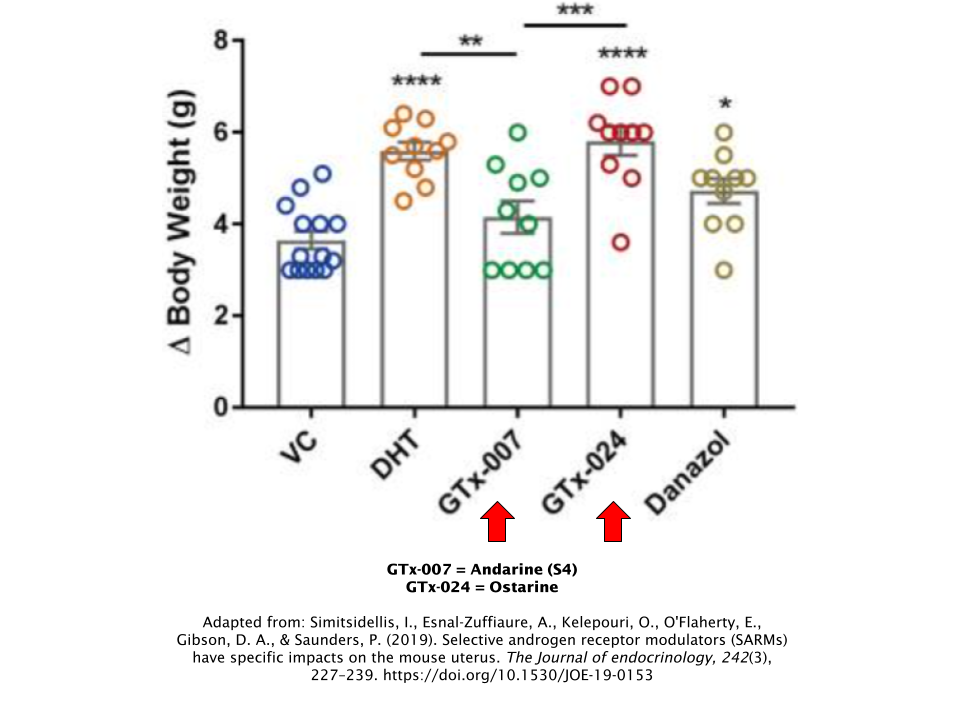
Ovariectomised mice (female mice that had their ovaries removed) were treated with either S4, Ostarine, DHT, or Danazol, the latter of which is a common drug used to treat women with breast cancer, muscle wasting, or urinary incontinence. Over the course of 1 week, Ostarine, DHT, and Danazol all succeeded in achieving the study objectives of increasing total body weight and uterus (uterine) weight, whereas S4 did not [7].
S23
Ironically enough, S23 is also a SARM developed by GTX Inc. S23 is currently being investigated for use as a hormonal male contraceptive, which should tell you it is significantly more suppressive of endogenous testosterone levels than S4.
The aspect of S23 that is responsible for its contraceptive effects in males is its ability to bind to the AR with significantly greater affinity (strength) compared to S4. S23 has shown to suppress spermatogenesis (the process during which sperm production originates) in a dose-dependent manner [8].
Data on S23 is quite limited, similar to S4. Given the dramatic suppressive effect on testosterone, it doesn’t seem to be advisable to use S23 in lieu of S4, despite S23’s greater anabolic potency. The only exception to this would be if an individual were to already be on a testosterone replacement therapy regimen alongside a cycle of S23.
Side Effects
Since there were no human trials conducted on S4, there’s not much data in which we can draw from in order to determine S4’s side effects on humans. However, there are some that have been reported anecdotally that should be taken note of.
Vision
This is perhaps the most infamous side effect of S4. The effect is often described by recreational users as a yellowish tint that occurs when switching from dark/dimly-lit environments to brighter areas.
While there is no official mechanistic explanation for this occurrence, it could be hypothesized that S4 binds to AR’s in the eye and its associated tissues. This may also be the reason why it was abandoned by researchers in favor of Ostarine.
Testosterone Suppression
Like any other SARM, S4 has the potential to shut down endogenous testosterone production. But it appears to be less suppressive than most other SARMs out there.
One study on castrated rats demonstrated that S4 suppressed LH (a key hormone for initiating the process of testosterone production), which indicates that S4 has the ability to shut down natural testosterone production. However, the dose used played a vital role in whether or not LH was inhibited in these rats. As a matter of fact, LH suppression wasn’t observed until a dose of 0.5mg/day or higher, which was far above the determined effective therapeutic dose for the rats [1].
This is illustrated in the figure below in the levator ani muscle, which again is that benchmark muscle in these animal studies that establishes the anabolic potency of SARMs. You’ll notice that the line begins to flatten out (indicating that the effective therapeutic dose has been reached) before the 0.5mg/day dose rate has been reached.
![]()
Assuming that this effect would translate into humans, we can hypothesize that there is minimal risk of testosterone shutdown at reasonable doses of S4.
S4 was also compared to DHT for its effects on LH and FSH and proved that it didn’t cause the same levels of suppression that DHT therapy did compared to the non-castrated control group.
![]()
Cholesterol
Cholesterol was not assessed in the animal studies conducted on S4, so it’s difficult to assert whether or not S4 would negatively affect cholesterol. But drawing upon what we know from previous SARMs data, we could err on the side of caution and assume that it will decrease HDL (“good”) cholesterol at the very least. But again, it should be noted that this has not been empirically established by any published research.
Recreational Doses & Cycles
Doses among recreational users greatly varies between one individual and the next. A brief look across the forums and message boards will tell you anecdotes of those who’ve done as little as 20mg/day all the way up to 100mg/day. And due to the fact that there is only preclinical data available for S4, no therapeutic dose was ever established either.
Although nobody knows exactly if the 4-hour half-life that was established in the animal studies would translate over to humans, it can be assumed that the half-life of S4 is significantly shorter in humans relative to the other SARMs that are out there. It may be wise to split your chosen dose into 2-3 smaller doses throughout the day rather than taking 1 larger dose, since this will create more even blood levels of the drug in your system throughout the day.
Bulking/Cutting Cycles
S4 is often compared to steroids that provide a hardening effect, such as Winstrol or Masteron. So S4’s most optimal use resides within a cutting/contest-prep cycle, but this isn’t to say that it couldn’t be used during a bulking phase as well. For either purpose, up to 100mg/day for 8 weeks would be sufficient.
During a bulking phase, It may be worth considering pairing S4 with other SARMs that are known for their muscle-building abilities, such as LGD-4033. As far as cutting phases go, SARMs like Cardarine and Ostarine are common SARMs to pair with S4.
Post-Cycle Therapy (PCT)
It’s highly recommended that you complete a PCT after an S4 cycle due to its ability to suppress total testosterone levels. The half-life of S4 is only ~4 hours according to the available preclinical data, therefore PCT should begin at the end of the same day you took your last dose.
It’s not necessary to include aromatase inhibitors in your PCT. Using a selective estrogen receptor modulator (SERM) should suffice; the two most popular choices being either tamoxifen (Nolvadex®) or clomiphene (Clomid®). While using tamoxifen (Nolvadex®) is recommended due to its milder nature, some prefer the use of clomiphene (Clomid®). Either way, one or the other should be just fine. Rarely is it necessary to take both of these SERMs simultaneously in order to recover properly.
No matter which one you choose, here’s what a simple 4-week PCT protocol would look like for these SERMS:
| Week | Tamoxifen (Nolvadex®) | OR | Clomiphene (Clomid®) |
|---|---|---|---|
| 1 | 40 mg/day | 50 mg/day | |
| 2 | 40 mg/day | 50 mg/day | |
| 3 | 20 mg/day | 25 mg/day | |
| 4 | 20 mg/day | 25 mg/day |
References
- Yin, D., Gao, W., Kearbey, J. D., Xu, H., Chung, K., He, Y., Marhefka, C. A., Veverka, K. A., Miller, D. D., & Dalton, J. T. (2003). Pharmacodynamics of selective androgen receptor modulators. The Journal of pharmacology and experimental therapeutics, 304(3), 1334–1340. https://doi.org/10.1124/jpet.102.040840
- NIDA. 2018, August 12. Anabolic Steroids DrugFacts. Retrieved from https://www.drugabuse.gov/publications/drugfacts/anabolic-steroids on 2021, August 7
- Kearbey, J. D., Wu, D., Gao, W., Miller, D. D., & Dalton, J. T. (2004). Pharmacokinetics of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro- 3-trifluoromethyl-phenyl)-propionamide in rats, a non-steroidal selective androgen receptor modulator. Xenobiotica; the fate of foreign compounds in biological systems, 34(3), 273–280. https://doi.org/10.1080/0049825041008962
- Mozayani. (2007). Handbook of drug interactions: A clinical and forensic guide.
- Kim, E. H., Larson, J. A., & Andriole, G. L. (2016). Management of Benign Prostatic Hyperplasia. Annual review of medicine, 67, 137–151. https://doi.org/10.1146/annurev-med-063014-123902
- Gao, W., Kearbey, J. D., Nair, V. A., Chung, K., Parlow, A. F., Miller, D. D., & Dalton, J. T. (2004). Comparison of the pharmacological effects of a novel selective androgen receptor modulator, the 5alpha-reductase inhibitor finasteride, and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia. Endocrinology, 145(12), 5420–5428. https://doi.org/10.1210/en.2004-0627
- Simitsidellis, I., Esnal-Zuffiaure, A., Kelepouri, O., O’Flaherty, E., Gibson, D. A., & Saunders, P. (2019). Selective androgen receptor modulators (SARMs) have specific impacts on the mouse uterus. The Journal of endocrinology, 242(3), 227–239. https://doi.org/10.1530/JOE-19-0153
- Jones, A., Chen, J., Hwang, D. J., Miller, D. D., & Dalton, J. T. (2009). Preclinical characterization of a (S)-N-(4-cyano-3-trifluoromethyl-phenyl)-3-(3-fluoro, 4-chlorophenoxy)-2-hydroxy-2-methyl-propanamide: a selective androgen receptor modulator for hormonal male contraception. Endocrinology, 150(1), 385–395. https://doi.org/10.1210/en.2008-0674